About Us (Group Leader)
- Norimichi Nomura Associate Professor
- Tatsuro Shimamura Project Lecturer
- Hidetsugu Asada Project Lecturer
- Makoto Adachi Assistant Professor
- Takuya Kobayashi
-------------------------------------------------------------------------------------------------------------------------------------------------------------
Generation and Crystallographic Analysis of Artificial Binder Molecules That Modulate
Membrane Protein Functions
Membrane proteins such as transporters, G-protein coupled receptors (GPCRs), channels, and ion pumps are attractive targets for development of
novel therapeutic drugs. Their X-ray crystal structures could potentially lead us to discover, design, and optimize more potent compounds with less serious
side effects for various disease conditions. However, such structure-guided drug design (SGDD) approaches remain stagnant, partly because it is still
challenging to achieve systematic crystallographic analyses of human integral membrane proteins.
The major impediment to membrane protein crystallography is production of high-quality diffracting crystals. For 3D crystallization trials,
membrane proteins are solubilized with detergents and purified as protein-detergent complexes. The detergent micelle covers the hydrophobic surface
(transmembrane regions) of the membrane proteins in a belt-like manner, and therefore, stable protein-protein contacts essential for crystal packing are not efficiently
formed. A strategy to overcome this problem is to use fragments of monoclonal antibodies, which specifically bind to a conformational epitope on the polar surface
of the membrane protein (See Figure). The artificially enlarged hydrophilic domain, with the aid of the bound antibody fragment, increases the probability of
obtaining well-ordered crystals. Our laboratory has established an efficient technological platform to generate suitable binding partners of human membrane proteins
for successful co-crystallization. Interestingly, a number of these binders also possess pharmacological activities to modulate the target membrane proteins.
Therefore, application of this strategy to a vast range of membrane proteins relevant to human pathogenesis should pave the way for SGDD, and the
“spin-off” binders might be utilized as promising candidates of therapeutics and/or diagnostics.
Our research efforts are focused on addressing the following two issues:
(i) Production and screening of artificial binder molecules that can act as a crystallization facilitator as well as a functional modulator
of membrane proteins. Molecular scaffolds of both antibody and non-antibody are to be employed.
(ii) X-ray crystallographic analyses of membrane proteins complexed with the functional binders to identify the pharmacophore structures
and to understand the molecular mechanisms of action as agonists or antagonists.
In conventional structural biology, a reductionist approach has prevailed that seeks to explain the biological significance of structures of individual molecules and complexes occurring in nature. Another new stream might emerge via our synthetic approach, in which molecular interactions between target membrane proteins and binders are rationally designed, controlled, and manipulated based on structural knowledge for engineering and directed modulation of cellular functions.
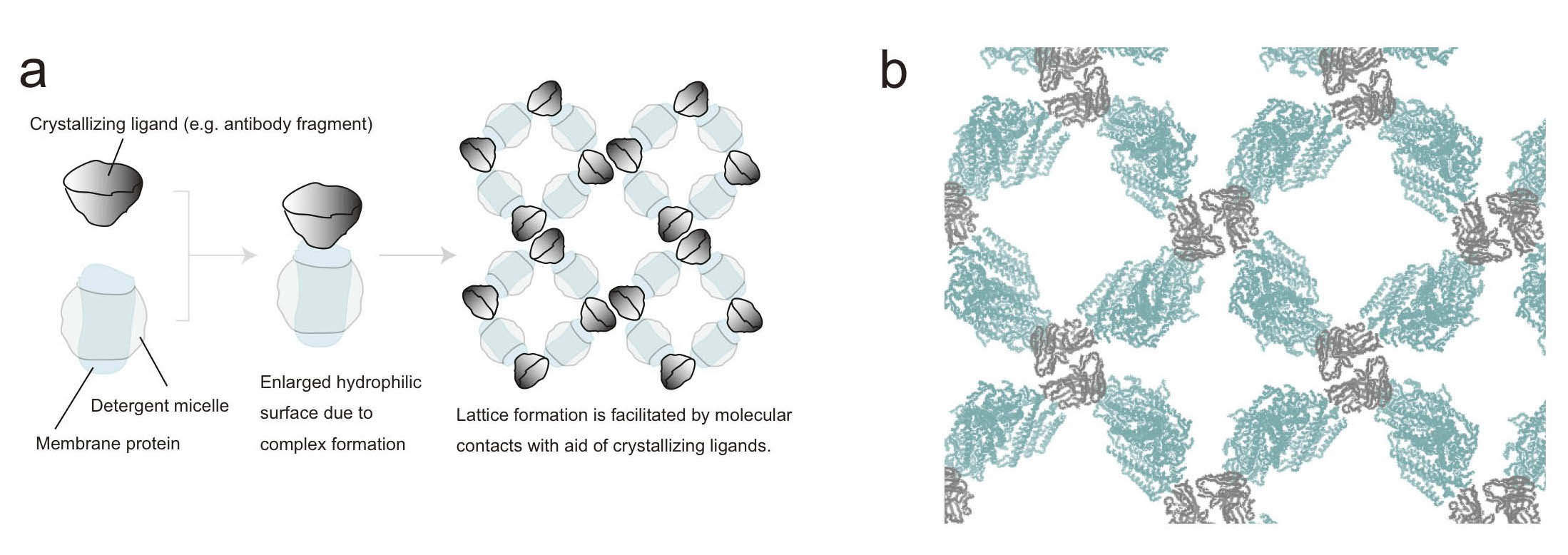
Figure. Antibody fragment-aided crystallization of membrane proteins
(a) Principle. A crystallizing ligand is an auxiliary protein that specifically binds to and forms stable complex with a target membrane protein of interest. The enlarged hydrophilic domain facilitates crystal lattice formation.
(b) An example of crystal packing of a membrane protein-antibody Fv fragment complex. The membrane protein and the Fv fragment are shown in light blue and gray, respectively. The Fv fragment contributes to crystal contacts and 3D crystal lattice formation.
-------------------------------------------------------------------------------------------------------------------------------------------------------------
Crystallographic studies of membrane proteins
Structure-based drug design is a useful tool for drug discovery. This method relies on the structural information of the drug targets. However,
~50% of the drug targets are membrane proteins whose structures are difficult to determine compared to soluble proteins.
Indeed, among ~94 000 structures in the Protein Data Bank, less than 1% of these structures represent membrane proteins, and the limitations of
membrane protein structure determination is a major bottleneck in the process of rational drug design.
Membrane proteins are difficult to crystallize because of various factors, including their hydrophobic nature, flexibility, and instability,
as well as the low abundance in native tissue. The ERATO Iwata human receptor crystallography project (2005~2011) studied mainly G-protein-coupled receptors (GPCRs).
During the project, we developed several techniques useful for the crystallization of membrane proteins. These techniques include overexpression systems of
GPCRs using Saccharomyces cerevisiae and Pichia pastoris, and an immunization and screening strategy to obtain monoclonal antibodies that recognize 3D structures
and stabilize the target protein. We also succeeded in determining the 3D structures of GPCRs by the lipidic cubic phase crystallization method.
In our group, we are trying to efficiently elucidate membrane protein structures by improving the techniques developed by the ERATO project and
by introducing new techniques through collaborative projects, such as those with SACLA, a Japanese X-ray Free Electron Laser facility (Fig. 1).
Through collaborative research, we are also conducting in silico screening studies, and computer simulation studies using the crystal structures of membrane proteins.
These studies are essential for the elucidation of the mechanisms of membrane proteins and rational drug discovery.
![]()
Figure1. Crystals of membrane proteins.
Structural analysis of drug target membrane proteins using XFEL
Our group is a member of the SACLA-SFX project, a MEXT project at SACLA, a Japanese X-ray Free Electron Laser (XFEL) facility.
XFEL provides highly intense and coherent X-ray pulses in durations of tens of femtoseconds. Therefore, using XFEL, we can avoid radiation damage to
protein crystals unlike with conventional X-ray sources.
The purpose of the SACLA-SFX project is to establish a strategy for the rapid structure determination of drug target membrane proteins at high resolution using SACLA. Riken, the research center of the project, is trying to develop new measurement equipment for data collection at SACLA. We are assisting Riken by providing protein nanocrystals and by collecting data at SACLA using the equipment. We also are establishing the overexpression systems of drug target membrane proteins and crystalizing the proteins for the structure determination using SACLA. For example, we have found that crystals of a membrane protein diffracts much better at SACLA (~1.9 angstrom) than at SPring-8 (~3.3 angstrom) (Fig. 2).
![]()
Figure2. Diffraction images. (A) SPring-8, (B) SACLA.
-------------------------------------------------------------------------------------------------------------------------------------------------------------
Structure-based drug design of GPCR (G protein-coupled receptor) by Sponsored Research Program
We are trying to elucidate the GPCR structure, in collaboration with Kyowa Hakko Kirin Co., Ltd. (KHK), in our research group by the Sponsored Research
Program (SRP). It is well known that about 30% of launched drugs target GPCRs. Therefore, GPCRs are considered very important membrane proteins. In this program,
our aim is to develop a novel drug based on the GPCR structure through a joint effort between industry and academia (Fig.).
In the present study, we are trying to elucidate the structure of GPCR using compounds at KHK. These compounds are in a developmental stage.
By elucidating the GPCR structure bound with the compound, we will be able to obtain important information for derivative screening of the compound.
This approach is called Structure-Based Drug Design (SBDD). It is thought that the information will contribute to the further development of SBDD.
In order for our project to succeed, we have to address the following difficult issues: 1) preparation of structurally stable mutants using a GFP-based
Saccharomyces cerevisiae screening system, 2) expression of a large amount of GPCRs using Sf9 insect cells and Pichia pastoris, 3) establishment of a
GPCR purification system, 4) preparation of a GPCR-specific structure recognition antibody, and 5) crystallization of GPCRs using the lipidic cubic phase method.
However, these procedures have already been established as fundamental technologies at the Kyoto site. Therefore, we will be able to elucidate the structure of target
GPCRs of KHK when we apply these technologies to this program.
One researcher from the Kyoto site, one or two researches from the KHK site, and two research assistants will take part in this program.
Both Kyoto and KHK sites are collaborating and supporting each other.
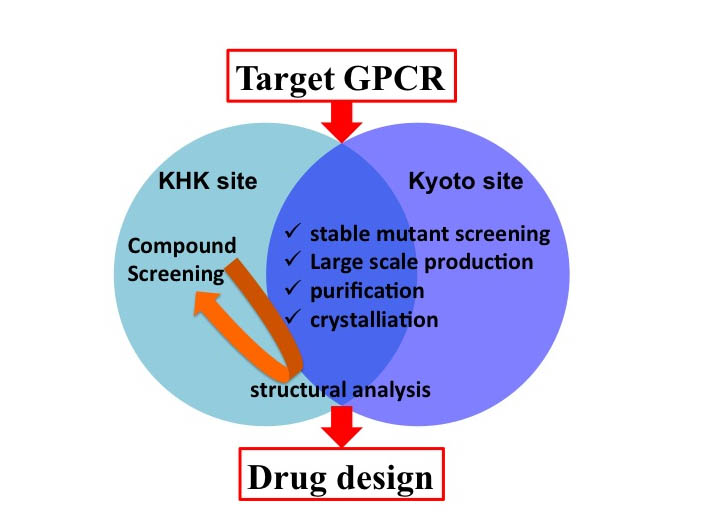
Figure. The schema of SRP study
The structural determination of target GPCRs is performed applying our fundamental technologies; the methods of stable transformants selection, large-scale production,
purification, and crystallization, at the Kyoto and KHK sites. The GPCR-specific compounds are improved at the KHK site.
-------------------------------------------------------------------------------------------------------------------------------------------------------------
Structure-based analysis of molecular functions of IQGAP family proteins
IQGAP family proteins are intracellular signaling molecules comprising IQGAP1, -2, and -3 in mammals, and possess the CH domain, WW domain, IQ domain,
and RasGAP-related domain (GRD) in this order from the NH2-terminus. Utilizing these domains, IQGAP proteins interact with a variety of molecules,
and are therefore involved in several functions. Functional abnormalities of these proteins are reportedly related to diseases, such as cancer.
Among the IQGAP family proteins, molecular functions of IQGAP3 are the least characterized. In previous studies, we have found that IQGAP3 is able to interact with,
and positively regulates the activity of, a small GTPase Ras. The IQGAP3-mediated regulation of Ras is important for proliferation of epithelial cells
and is achieved via the GRD of IQGAP3. Importantly, although GRD generally acts as a negative regulator of small GTPases, GRD of IQGAP3 rather acts as a
positive regulator of Ras. Thus, the GRD of IQGAP3 possesses a property opposite to conventional GRDs. To understand the molecular mechanisms underlying
this unique property, it is very important to elucidate its three-dimensional structure. We are therefore performing a crystal structure analysis utilizing
recombinant proteins of the GRD of IQGAP3.
In this way, our goal is to find interesting functions of proteins applying cell biological or biochemical studies, and then clarify the responsible
molecular mechanisms at the structural level by a crystal structure analysis. In our recent study, an IQGAP protein was found to interact with a functional
protein to achieve a novel function other than that described above. We are now analyzing the regulation and detailed functions of this protein complex and,
in parallel, expressing and purifying their recombinant proteins to perform a crystal structure analysis.
Through these studies, we want to clarify the molecular functions of IQGAP proteins from a variety of viewpoints.
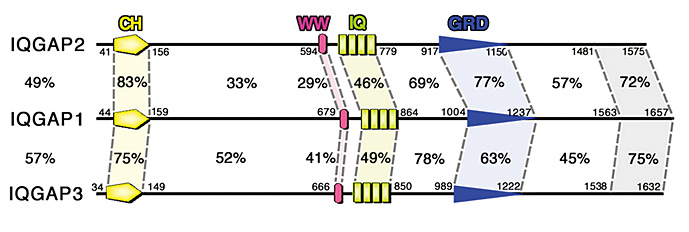
Figure. A schematic diagram of mammalian (mouse) IQGAP family proteins.
-------------------------------------------------------------------------------------------------------------------------------------------------------------
Towards structure-based GPCR pharmacology: from knockout mice to crystal structures
Recent successes in the high-resolution determination of G-protein coupled receptor (GPCR) structures have demonstrated the importance of receptor stability.
The rapid screening and monitoring of functionally expressed stable receptors are vital for structural studies.
The present study proposes the use of a GFP-based Saccharomyces cerevisiae membrane protein over-expression system as an initial host for the rapid construction
and evaluation of functional GPCR variants for structural studies (Proc. Natl. Acad. Sci. USA 104: 13936-139341, 2007, Nat. Protoc. 3: 784-798, 2008,Microb. Cell Fact. 11:78, 2012).
The GPCR variants displayed markedly improved functional activity in S. cerevisiae and monodispersity by fluorescence size exclusion chromatography (FSEC).
The improved GPCR variants correlated well with those functionally expressed in either P. pastoris or Sf9 insect cells, and much higher expression levels were achieved in these hosts.
Using this system, the expression level of the human histamine H1 receptor (HRH1) variant in P. pastoris has achieved 650-fold increases over that of the wild type HRH1 in S. cerevisiae.
We have determined the crystal structure of the human HRH1 at 3.5 angstrom (Nature 475: 65-70, 2011). Thus, this GFP-based S. cerevisiae system may serve as a powerful platform for the construction and selection of stable GPCR variants.
We have reported the raising of a mouse monoclonal antibody against a human adenosine A2a receptor that prevents agonist but not antagonist binding to the extracellular ligand-binding pocket, and described the structure of the adenosine A2a receptor in complex with the antibody Fab fragment (Fab2838) (Nature 482: 547-551, 2012). This structure revealed that Fab2838 recognizes the intracellular surface of the adenosine A2a receptor and that its complementarity-determining region, CDR-H3, penetrates into the receptor. CDR-H3 is located in a similar position as the G-protein carboxy-terminal fragment in the active opsin structure and as CDR-3 of the nanobody in the active β2-adrenergic receptor structure, but locks the adenosine A2a receptor in an inactive conformation. These results suggest a new strategy to modulate the activity of G-protein-coupled receptors.
We collaborated with Prof. Tatsuya Haga and Prof. Brian Kobilka and recently reported on the M2 muscarinic acetylcholine receptor (M2 receptor) (Nature 482: 547-551, 2012). Muscarinic receptors are G-protein-coupled receptors that mediate the response to acetylcholine released from parasympathetic nerves. M2 receptor is of particular interest because of its extensive pharmacological characterization of both orthosteric and allosteric ligands. We have elucidated the structure of the antagonist-bound human M2 receptor, the first human acetylcholine receptor to be characterized structurally. The antagonist 3-quinuclidinyl-benzilate binds in the middle of a long aqueous channel extending approximately two-thirds through the membrane. The orthosteric binding pocket is formed by amino acids that are identical in all five muscarinic receptor subtypes. A layer of tyrosine residues forms an aromatic cap restricting dissociation of the bound ligand. A binding site for allosteric ligands has been mapped to residues at the entrance to the binding pocket near this aromatic cap. The structure of the M2 receptor provides insights into the challenges of developing subtype-selective ligands for muscarinic receptors and their propensity for allosteric regulation.
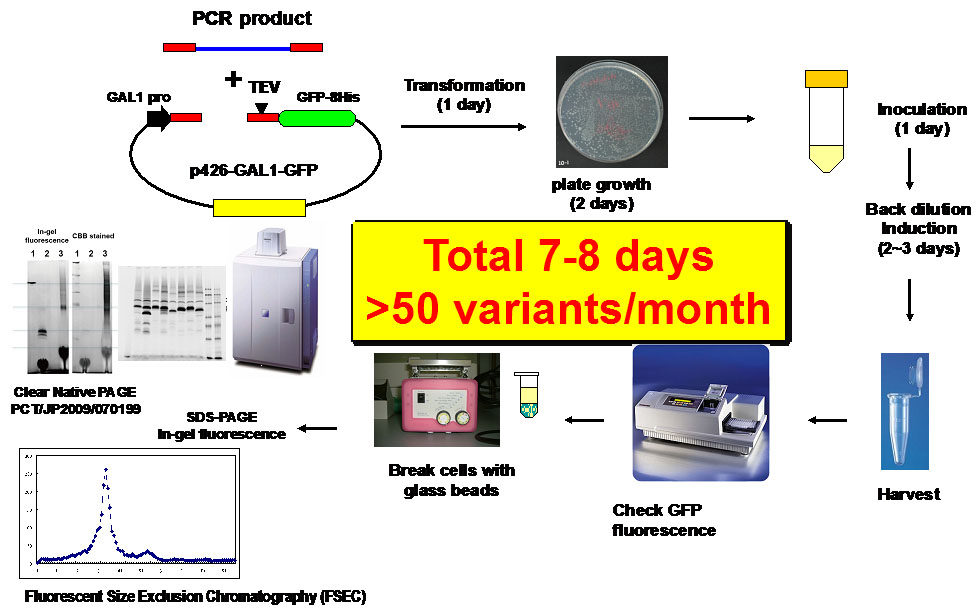
Figure1. Flow chart of a GFP-based Saccharomyces cerevisiae membrane protein over-expression system.
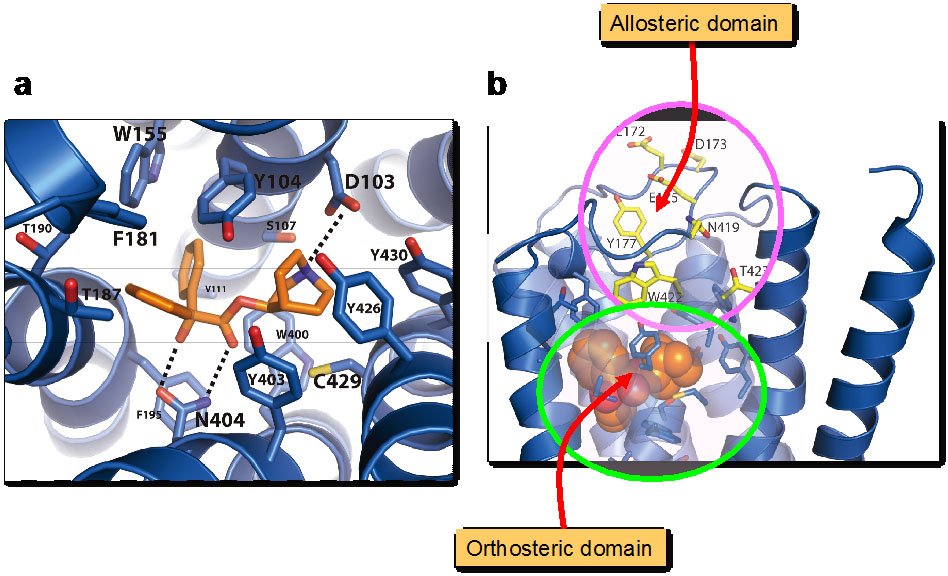
Figure2. Structure of muscarinic M2 receptor.
a) QNB bound orthosteric domain.
b) Allosteric domain lining the path to the QNB binding pocket.